What Is the Difference Between Qualification and Mapping?
For temperature-controlled equipment and areas, mapping analysis forms part of the qualification plan. Often mapping is carried out during multiple stages of the qualification process— typically during IQ, OQ, and PQ phases. Mapping is the examination and evaluation part of qualification, providing you with the data for trends, simulated failures, and thermal reaction times to assess. Mapping during qualification often involves varied product loads inside the chamber under study, starting off empty and moving towards a typical operational load.
A mapping study defines the behavioral pattern within the room, while the qualification study follows through with a detailed analysis to show the design and operations match the expected results.
What Are Formal Requirements for a Qualification?
There are no exact formal requirements. So, whosoever wants to perform the process is free to choose what form and document structure to use. However, you should observe good documentation practices. Use clear document naming, versioning, and change the history and ownership of the document.
Why and What to Qualify
According to GxP validation guidelines, you must qualify all equipment impacting a patient's safety and authenticate its processes.
Some of the relevant equipment includes:
- Temperature monitoring equipment
- Containers and transport boxes
- Trucks or van fleets
- Warehouses
- Networks
Qualification and Mapping Examples
Refrigerators and Freezers
Qualifying temperature-controlled refrigerators involves risk-based testing for airflow, loading, and door openings. Mapping ensures regulatory compliance and ongoing data integrity.
Warehouses and Cold Rooms
Large storage facilities require seasonal temperature mapping due to ambient influences. Cold rooms often require less frequent mapping but still demand validation for GxP compliance.
Transport Containers and Trucks
Transport qualification ensures temperature-sensitive goods remain within range during transit. Both passive insulated boxes and active containers must be qualified and mapped to meet Good Distribution Practice and CFR Part 11 requirements.
How to Conduct a Qualification and Mapping
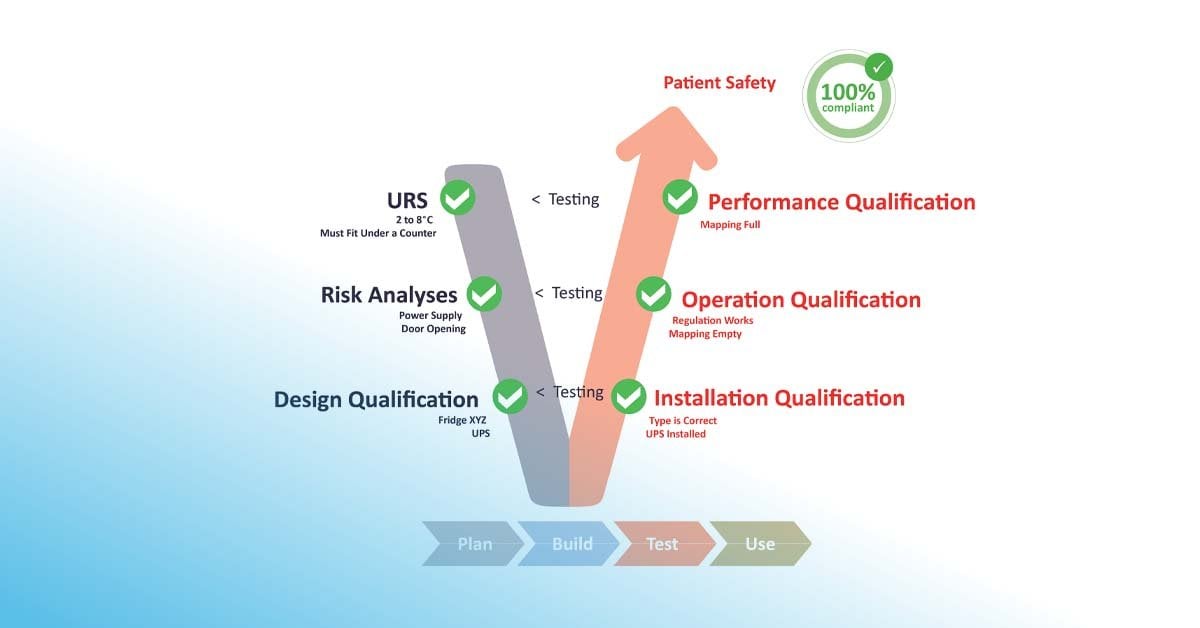
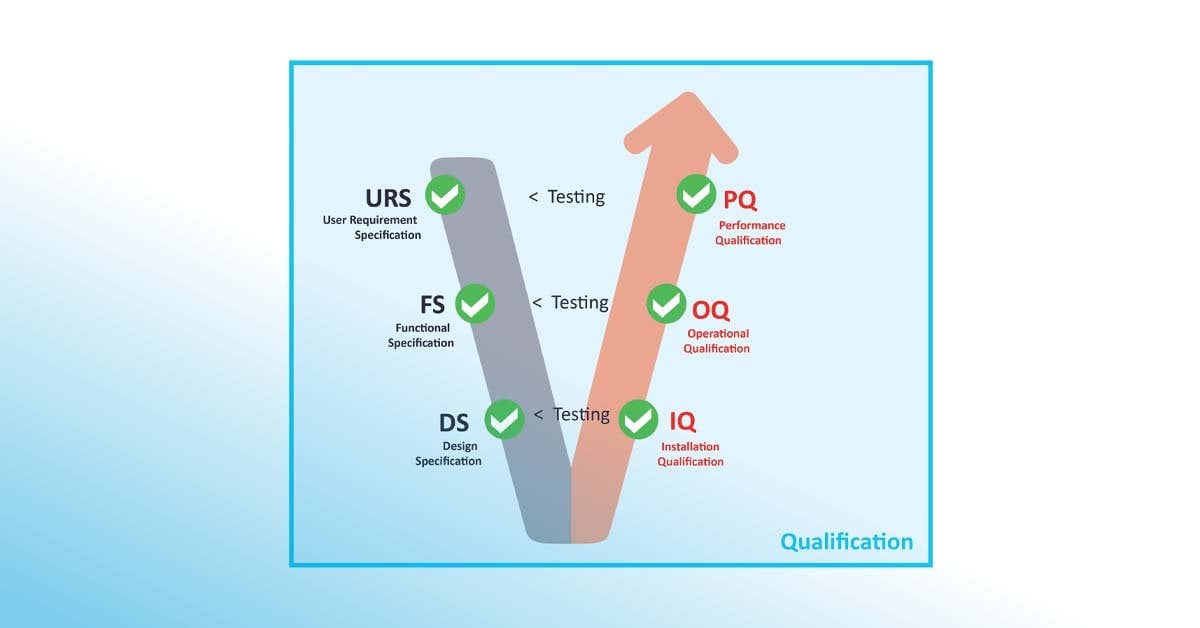
In the pharmaceutical sector, the qualifying process differs depending on the equipment and space. In some instances, specialized engineering firms conduct the qualification. ELPRO uses the GAMP® 5 V model as a guide, the steps 3, 4, 5, and 6 being the qualification portion.
- Define the purpose of your facility – is it a warehouse or a cold room?
- Perform an analysis of structural and operational risks.
- Design Qualification: Define high-level design requirements, like power supply and floor plans.
- Installation Qualification: Check to establish if the facility is built as designed.
- Operation Qualification: Conduct an operational qualification by mapping the facility when empty.
- Performance Qualification: Perform the second temperature mapping during normal operations with a full load.
Here are some valuable examples of how to conduct qualification and temperature mapping in various circumstances:
How to Qualify and Map a Refrigerator
Qualifying a refrigerator is straightforward because it is a freestanding gadget. Since these appliances operate in a room or environment with controlled temperature, risks are limited to air outlet blockage, power outage, improper loading, and door opening. In comparison to more intricate devices and personalized equipment, there are minimal risks.
Companies usually operate numerous refrigerators or freezers, so the qualifying processes are often done in the following steps:
- Assemble the refrigerators into homogenous categories under the same supplier, model, and size.
- Apply the V model to each group.
- Perform the design check for a single refrigerator per cluster group by confirming its model, airflow, and power supply specifications.
- After mapping out a single refrigerator in each group, the remainder can skip the step and proceed to performance qualification.

/Qualification%20and%20Mapping/Risk%20Strategy.png?width=855&name=Risk%20Strategy.png)