GxP Compliant Monitoring
Pharma GxP for Beginners
Getting started within the Pharma GxP can be quite overwhelming. Most important is to get an overview of what GxP is all about. This article gives...
You are not sure which GxP regulations applies for Germany and Switzerland in the Pharmaceutical Industry? This article will answer all you need to know about it.
Individual market participants who handle medicinal products and medical devices commercially face the question of which relevant standards, laws, regulations, directives, etc., must be observed and complied with in individual cases and on account of the segment in the supply chain.
It's important to know who the controlling bodies are, the standard regulations, the important terms, and when is which calibration required.
In Germany, federal authorities carry out approval-related inspections in this regard. In addition, only the competent authority of the respective federal state is on site as a rule, i.e., regional authorities, district governments, regional directorates, state ministries in the respective federal states, and carries out appropriate inspections and takes action if they discover violations of the provisions.
In addition, the authorities are responsible for health protection (including monitoring through inspections according to the GMP, GDP, etc.). Their responsibilities are regulated differently in the federal states. Information on which authority performs which tasks in which federal state can be found in the directory of authorities, agencies, and experts responsible for the enforcement of the German Medicinal Products Act
The Central Authority of the Länder for Health Protection (ZLG), based in Bonn, has an important coordinating function. The ZLG handles the consistency of monitoring, coordinates interstate measures and inspections in the central approval procedure, and exchanges information with European and international monitoring institutions. It also coordinates the activities of the medicinal product investigation agencies in the federal states (Länder).
Swissmedic is the central Swiss supervisory authority for therapeutic products. As a federal institution under public law with its headquarters in Bern, it is independent in its organization and management and has its own budget. Swissmedic itself only handles the controlling of blood and rare procedures, but it remains responsible for authorizations and operating licenses.
Therapeutic Products Control:
The cantonal inspectorates are responsible for general GMP/GDP inspections. They are supervised and coordinated by Swissmedic. The following inspectorates exist in Switzerland:
Regional Medicines Inspectorate of Northwestern Switzerland (Regionales Heilmittelinspektorat der Nordwestschweiz, RHI NW)
Regional Medicines Inspectorate of Eastern and Central Switzerland (Regionale Fachstelle der Ost- und Zentralschweiz, RFS-OZ)
Regional Medicines Inspectorate of Southern Switzerland (Regionales Medizin-Inspektorat der Südschweiz, IRM-S)
The Food and Drug Administration (FDA) is the U.S. food and drug regulatory agency. The FDA and other foreign authorities may be authorized by local authorities (e.g., Swissmedic) to inspect Swiss companies.
GxP is the abbreviation for general good practice, which refers to a set of laws, provisions, and guidelines that govern various areas of research, development, testing, manufacturing, and distribution of medicinal products. The “x” can be substituted by the letters explained below.
Good laboratory practice (GLP) refers to guidelines for the quality assurance of the framework conditions and organizational procedures under which non-clinical health and environmental safety audits are planned, performed, and monitored. This also includes the recording, archiving, and reporting of audits.
Good manufacturing practice (GMP) refers to guidelines for quality assurance of the production environment and production processes in the manufacturing of medicinal products and active pharmaceutical ingredients, as well as cosmetics, food, and feed.
Good distribution practice refers to guidelines for quality assurance in the complete supply chain of active pharmaceutical ingredients and medicinal products.
The pharmacopoeia is published by the EU as the European Pharmacopoeia (Pharmacopoeia Europaea – Ph. Eur.) and contains quality provisions for common and known medicinal products and pharmaceutical excipients that are risk-appropriate and based on the state of the art in science and technology. The provisions contained in it are binding and have force of law. The pharmacopoeia thus makes a significant contribution to ensuring that therapeutic products of the same high quality are available to all patients and thus creates a central prerequisite for safe and effective therapeutic products.
The Swiss Pharmacopoeia (Pharmacopoea Helvetica – Ph. Helv.) consists of the European Pharmacopoeia and the Swiss Pharmacopoeia. It was issued by Swissmedic on the basis of the Swiss Therapeutic Products Act (TPA); it contains quality requirements in line with the level of risk and based on the state of the art in science and technology for customary and known medicinal products, pharmaceutical excipients, and individual medical devices.
This international standard specifies the general requirements for competence in the performance of tests and/or calibrations, including sampling. ISO 17025 accreditation means that the testing and/or calibration laboratories are technically competent and capable of producing professionally sound results. Accreditations according to ISO 17025 are carried out centrally in Switzerland by the Swiss Accreditation Service SAS.

ISO 13485 is an ISO standard that represents the requirements for a comprehensive quality management system for the design and manufacture of medical devices. The standard contains detailed requirements on topics concerning the design, manufacture, and placing on the market of medical devices. Compliance with the standard is proven by accreditation and continuously monitored.
The good automated manufacturing practice (GAMP) supplier guide for validation of automated systems in pharmaceutical manufacture was published in collaboration with the International Society for Pharmaceutical Engineering (ISPE). The GAMP guide is intended as a guideline without a legally binding character; nevertheless, it has become the standard set of rules for the validation of computerized systems (CSV) in the pharmaceutical industry (manufacturers and suppliers). Regarding GAMP 5, the ISPE website states: “GAMP 5: A Risk-Based Approach to Compliant GxP Computerized Systems provides pragmatic and practical industry guidance that aims to achieve compliant computerized systems that are fit for intended use in an efficient and effective manner, while also enabling innovation and technological advances.” [URL: https://ispe.org/product-types/gamp-good-practice-guides]
FDA 21 Code of Federal Regulation, Part 11 (FDA 21 CFR Part 11) is a law of the United States of America. In it, the FDA formulates requirements for electronic records and signatures directed at pharmaceutical and medical device manufacturers. FDA 21 CFR Part 11 applies whenever information is to be created, modified, stored, transmitted, or accessed electronically.
The International Air Transport Association (IATA) was founded as the umbrella organization for airlines. The aim of the IATA is, among other things, to promote the safe, scheduled, and economical transport of people and goods by air – which implicitly includes pharmaceuticals and medical devices.
Validation provides documented proof that a process or system reproducibly meets the previously specified requirements (acceptance criteria) in practical use. A prerequisite for such a qualification is typically a validation of the associated process. In order to validate a transport process, for example, the vehicles, temperature sensors, employees, etc., used in the process must also be appropriately qualified.
During the qualification of skilled personnel, instruments, and facilities, their suitability for the intended task with the technology used is checked. A qualification is therefore documented proof that a room, a system, a facility, a supplier, or an employee is suitable for the intended purpose, or two or more are suitable in interaction. If physical parameters are also measured, calibration is a prerequisite for qualification.
There is a rough rule of thumb for differentiating between validating and qualifying: What you can touch is qualified, what you can’t touch is validated.
A testing or calibration laboratory is accredited if, for example, it meets the requirements of the DIN EN ISO/IEC 17025 standard. For this purpose, the laboratory is assessed by assessors from an independent accreditation body that complies with the DIN EN ISO/IEC 17011 standard and is monitored through assessments at intervals of one to one and a half years.
Certification is a process used to demonstrate compliance with certain requirements. Certification is a sub-process of conformity assessment. Certifications are often issued by independent certification bodies for a limited period of time; for example, a certificate limited to one year is issued for a temperature sensor.
Calibration in metrology is a measurement process for reliably reproducible determination and documentation of the deviation of a measuring instrument or a material measure from another instrument or another material measure, which is referred to as a standard in this case.
During adjustment, an instrument is technically modified in order to achieve the lowest possible deviation, for example of the temperature sensor. Calibration must always be performed after adjustment.
Many experts working in the pharmaceutical and medical devices sector use acronyms familiar to them to designate processes, provisions, and instances. A selection of relevant acronyms commonly used in this field can be found in the table below:
| Abbreviation | English term | German term |
| API | Active pharmaceutical ingredient | Pharmazeutischer Wirkstoff |
| CAPA | Corrective and preventive action | Korrektur- und Vorbeugemassnahme |
| FDA | Food and Drug Administration | US Lebensmittel- und Arzneimittelbehörde |
| GAMP | Good automated manufacturing practice | Gute automatisierte Fertigungspraxis |
| GCP | Good clinical practice | Gute klinische Praxis (klinische Studien) |
| GDP | Good distribution practice | Gute Vertriebspraktiken |
| GLP | Good laboratory practice | Gute Laborpraxis |
| GMP | Good manufacturing practice | Gute Herstellungspraxis |
| IATA | International Air Transport Association | Internationaler Luftfahrtverband |
| IMP | Investigational medicinal products | Arzneimittel für eine klinische Prüfung |
| ISO | International Organization for Standardization | Internationale Organisation für Normung |
| ISPE | International Society for Pharmaceutical Engineering | Intern. Gesellschaft für Pharmaz.Technik |
| PIC | Pharmaceutical Inspection Convention | Pharmazeutische Inspektionskonvention |
| PiT | Product in transit | Produkt im Transport |
| SAS | Swiss Accreditation Service | Schweizerische Akkreditierungsstelle |
| SCS | Swiss Calibration Service | Schweizerischer Kalibrierdienst |
| SW | Software | Software |
The following table indicates which type of calibration is required or expected by the authorities in which area of the pharmaceutical supply chain.
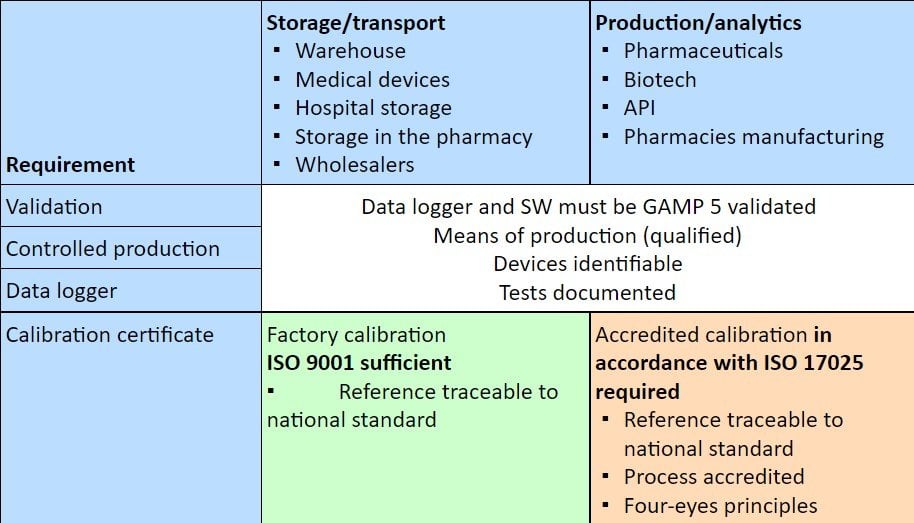
The catalog of requirements for handling pharmaceutical products and medical devices throughout the supply chain is diverse and may appear to some as a jungle of standards. The danger of overlooking or forgetting something is ever-present. Especially in the area of sufficient calibration, there are more and more questions as to what the authorities rightly require and what actually an overreach is. Of course, the requirements in the various laws, directives, regulations, etc., are binding and therefore mandatory.
Getting started within the Pharma GxP can be quite overwhelming. Most important is to get an overview of what GxP is all about. This article gives...
What does a temperature mapping study tell you? How does documentation affect audits? ELPRO's GxP expert answer your most common mapping questions.
The basic requirements for accurate temperature monitoring.